| WGS全基因组测序数据分析教程(一) | 您所在的位置:网站首页 › hla基因测序 › WGS全基因组测序数据分析教程(一) |
WGS全基因组测序数据分析教程(一)
|
一、全基因组测序(WGS) 的定义 全基因组测序(WGS, Whole Genome Sequencing)是下一代测序技术,用于快速,低成本地确定生物体的完整基因组序列。其目的是准确检测出每个样本(这里特指人)基因组中的变异集合,也就是人与人之间存在差异的那些DNA序列。 生物的主要遗传物质是DNA,细胞或生物体中一套完整的遗传物质的总和称为基因组。DNA是由外显子(Exon)和内含子(Intron)组成。外显子只占基因组的1%左右,指导体内所有蛋白的合成。内含子不编码蛋白质的合成,但绝不是无用的序列,其可以影响基因的活性和蛋白的表达。全基因组测序,即是对生物体整个基因组序列进行测序,可以获得完整的基因组信息。 三个需要区分的概念:WES (whole exome sequencing, 全外显子组测序): 是指利用序列捕获技术将【全基因组的外显子区域DNA】捕获富集后进行高通量测序,能够直接发现与蛋白质功能变异相关的遗传变异SNP(单核苷酸多态性)。以人类基因组为例,虽然外显子(蛋白质编码区)只占基因组的1%,但人类基因组85%的致病突变都在外显子区域,因此具有重要意义。WES仅是对外显子区域的测序。相比全基因组测序,外显子测序更为简便,测序成本相比也会更低,测序后数据的分析也更为简单。另外如果仅关注已知的基因,可以更依赖于靶向测序。 WGS (whole genome sequencing, 全基因组测序): 是指对基因组整体进行高通量测序,分析不同个体间的差异,同时完成SNP及基因组结构注释。全基因组测序由于结果包含完整丰富的信息,可以得到外显子测序或靶向测序不能得到的更多信息,具有其独特的优势。且随着近年来测序技术的不断进步、测序成本的不断降低,使得全基因组测序变得触手可及。而且全基因组测序在鉴定单核苷酸变异(SNP)、插入和缺失突变(Indel)时更有优势,所以WGS逐渐成为了临床和基础研究的另一种选择。 WGRS (whole genome re-sequencing, 全基因组重测序): 是指对已知参考基因组和注释的物种进行不同个体间的全基因组测序(WGS),并在此基础上对个体或群体进行差异性分析,鉴定出与某类表型相关的SNP。 三个技术的覆盖程度不同:① WES:覆盖全基因组上的外显子区域 ② WGS:覆盖全基因组 ③ WGRS:覆盖全基因组,是WGS在不同样本上的重复 三种主要变异分类:① 单核苷酸多态性(SNP):单碱基变异,最常见也最简单的一 种基因组变异形式; ② 插入和缺失(Indel):很短的Insertion 和 Deletion,也常被我们合并起来称为Indel。主要指在基因组某个位置上发生较短长度的线性片段插入或者删除的现象。强调线性的原因是,这里的插入和删除是有前后顺序的与下述的结构性变异不同。Indel长度通常在50bp以下,更多时候甚至是不超过10bp,这个长度范围内的序列变化可以通过Smith-Waterman 的局部比对算法来准确获得,并且也能够在目前短读长的测序数据中较好地检测出来; ③ 基因组结构性变异(Structure Variantions,简称SVs),这篇文章的重点,通常就是指基因组上大长度的序列变化和位置关系变化。类型很多,包括长度在50bp以上的长片段序列插入或者删除(Big Indel)、串联重复(Tandem repeate)、染色体倒位(Inversion)、染色体内部或染色体之间的序列易位(Translocation)、拷贝数变异(CNV)以及形式更为复杂的嵌合性变异。也有将拷贝数变异(CNV)单独将作为第四类变异。 二、WGS分析流程(以重测序为例) 三、分析工具安装准备 三、分析工具安装准备① BWA(Burrow-Wheeler Aligner)使用最广的NGS数据比对软件;适用范围:将短序列拼接至模板基因组。 ② Samtools:是一个专门用于处理比对数据的工具,由BWA的作者所编写; ③ Picard:也是一款强大的NGS数据处理工具,功能方面和samtools有些重叠,但更多的是互补,它是由java编写的,我们直接下载最新的.jar包就行了。 ⑤ GATK:是目前业内最权威、使用最广的基因数据变异检测工具。 ⑥ SnpEff:是一款用于注释变异(SNP、Small InDel)和预测变异影响的软件。 ⑦ 结构变异(SVs/CNV)检测工具:这种软件相对比较多,都有适用的结构变异类型,如:pindel,delly,breakdancer,Control-FREEC等。 四、数据质控数据错误率的来源:以illumina为例目前的测序技术基本都是运用边合成边测序的技术,碱基的合成依靠的是化学反应,这使得碱基链可以不断地从5'端一直往3'端合成并延伸下去。但在这个合成的过程中随着合成链的增长,DNA聚合酶的效率会不断下降,特异性也开始变差,这会导致越到后面碱基合成的错误率就会越高,这也是为何当前NGS(下一代测序)测序读长普遍偏短的一个原因。 认识一个数据好坏从以下方面入手:可以通过一些工具或者R代码来展示数据质量 read各个位置的碱基质量值分布。碱基的总体质量值分布。read各个位置上碱基分布比例,目的是为了分析碱基的分离程度。GC含量分布。read各位置的N含量。read是否还包含测序的接头序列。read重复率,这个是实验的扩增过程所引入的。质控工具:Trimmomatic(java语言运行速度慢)和fastp(快)软件具体用法介绍可以参考我前面分享内容 五、数据比对与预处理先问一个问题:为什么需要比对? 我们已经知道NGS测序下来的短序列(read)存储于FASTQ文件里面。虽然它们原本都来自于有序的基因组,但在经过DNA建库和测序之后,文件中不同read之间的前后顺序关系就已经全部丢失了。因此,FASTQ文件中紧挨着的两条read之间没有任何位置关系,它们都是随机来自于原本基因组中某个位置的短序列而已。 因此,我们需要先把这一大堆的短序列捋顺,一个个去跟该物种的参考基因组*比较,找到每一条read在参考基因组上的位置,然后按顺序排列好,这个过程就称为测序数据的比对。这也是核心流程真正意义上的第一步,只有完成了这个序列比对我们才有下一步的数据分析。 序列比对本质上是一个寻找最大公共子字符串的过程。大家如果有学过生物信息学的话,应该或多或少知道BLAST,它使用的是动态规划的算法来寻找这样的子串,但在面对巨量的短序列数据时,类似BLAST这样的软件实在太慢了!因此,需要更加有效的数据结构和相应的算法来完成这个搜索定位的任务。我们这里将用于流程构建的BWA就是其中最优秀的一个,它将BW(Burrows-Wheeler)压缩算法和后缀树相结合,能够让我们以较小的时间和空间代价,获得准确的序列比对结果。 1. 构建基因组索引首先,我们需要为参考基因组的构建索引——这其实是在为参考序列进行Burrows Wheeler变换(wiki: 块排序压缩),以便能够在序列比对的时候进行快速的搜索和定位。 $ bwa index human.fasta以我们人类的参考基因组为例,这个构造过程需要一段时间。完成之后,你会看到类似如下几个以human.fasta为前缀的文件,这一步完成之后,我们就可以将read比对至参考基因组了: . ├── human.fasta.amb ├── human.fasta.ann ├── human.fasta.bwt ├── human.fasta.pac └── human.fasta.sa2. 比对# bwa mem -t 4 human.fasta read_1.fq.gz read_2.fq.gz > sample_name.sam #一般不生成sam文件,文件太大占用空间, # 为了有效节省磁盘空间,一般都会用samtools将它转化为BAM文件(SAM的特殊二进制格式) $ bwa mem -t 4 human.fasta read_1.fq.gz read_2.fq.gz | samtools view -S -b - > sample_name.bam我们通过管道(“|”)把比对的输出如同引导水流一样导流给samtools去处理,上面samtools view的-t 表示线程数,-b参数指的就是输出为BAM文件,这里需要注意的地方是-b后面的 '-',它代表就是上面管道引流过来的数据,经过samtools转换之后我们再重定向为sample_name.bam 3. 排序为什么需要排序?因为BWA比对后输出的BAM文件是没顺序的,每一条记录之间位置的先后顺序是乱的,我们后续去重复等步骤都需要在比对记录按照顺序从小到大排序下来才能进行,所以这才是需要进行排序的原因。这个排序的命令如下: # -@,用于设定排序时的线程数,我们设为4;-m,限制排序时最大的内存消耗,这里设为4GB;-O 指定输出为bam格式 # 排序后如果发现新的BAM文件比原来的BAM文件稍微小一些,很正常,这是压缩算法导致的结果,文件内容是没有损失的。 $ bwa mem -t 4 human.fasta read_1.fq.gz read_2.fq.gz | samtools view -S -b - |samtools sort -o sample_name.sort.bam4. 下游比对数据的统计4.1 比对结果统计 与参考基因组比对,统计样品比对率等信息.比对率:即可以定位到参考基因组上的Clean Reads占总的Clean Reads数比例,如果参考基因组选择合适,且相关实验过程不存在污染,测序reads的比对率会高于70%。另外,比对率的高低受测序物种与参考基因组亲缘关系远近、参考基因组组装质量高低及reads测序质量有关,物种越近缘、参考基因组组装越完整、测序reads质量越高,则可以定位到参考基因组的reads也越多,比对率越高。 工具:samtools flagstat命令实现 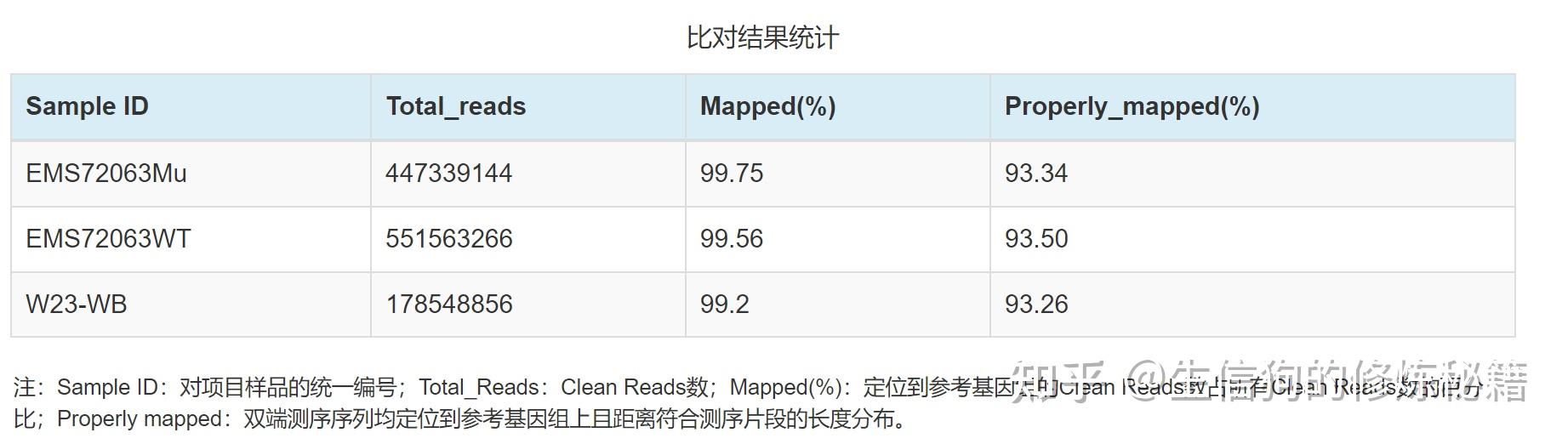 4.2 插入片段分布统计 4.2 插入片段分布统计通过检测双端序列在参考基因组上的起止位置,可以得到样品DNA打断后得到的测序片段的实际大小,即插入片段大小(Insert Size),它是信息分析时的一个重要参数。插入片段大小的分布一般符合正态分布,且只有一个单峰,Insert Size分布图可以展示各个样品的插入片段的长度分布情况。 工具:picard软件工具包中CollectInsertSizeMetric.jar软件实现。 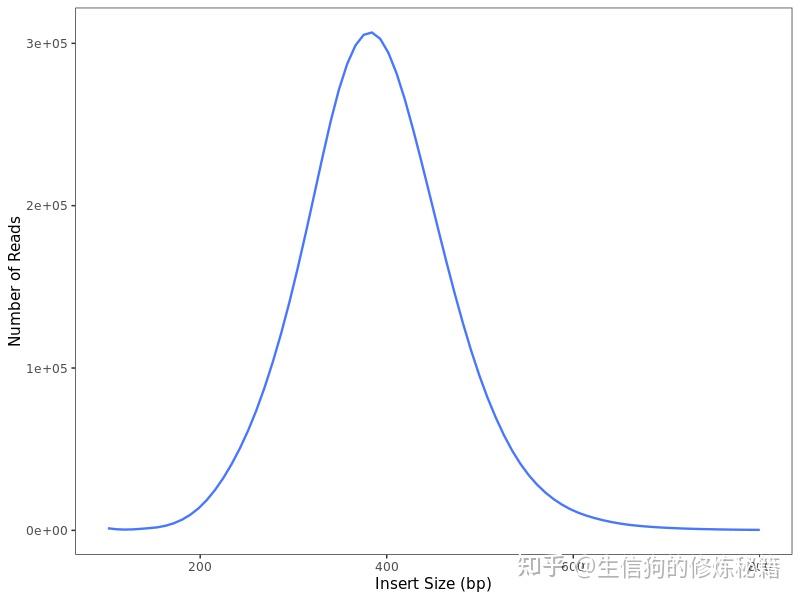 注:横坐标为插入片段长度,纵坐标为其对应的reads数4.3 深度分布统计 注:横坐标为插入片段长度,纵坐标为其对应的reads数4.3 深度分布统计Reads定位到参考基因组后,可以统计参考基因组上碱基的覆盖情况。参考基因组上被reads覆盖到的碱基数占基因组的百分比称为基因组覆盖度;碱基上覆盖的reads数为覆盖深度。基因组覆盖度可以反映参考基因组上变异检测的完整性,覆盖到的区域越多,可以检测到的变异位点也越多。覆盖度主要受测序深度以及样品与参考基因组亲缘关系远近的影响。基因组的覆盖深度会影响变异检测的准确性,在覆盖深度较高的区域(非重复序列区),变异检测的准确性也越高。另外,若基因组上碱基的覆盖深度分布较均匀,也说明测序随机性较好。样品的碱基覆盖深度分布曲线和覆盖度分布曲线见下图:工具:samtools depth命令  注:上图反映了测序深度分布的基本情况,横坐标为测序深度,左纵坐标为该深度对应的碱基所占百分比,对应红色曲线,右纵坐标为该深度及以下的碱基所占百分比,对应蓝色曲线。4.4 染色体覆盖度统计 注:上图反映了测序深度分布的基本情况,横坐标为测序深度,左纵坐标为该深度对应的碱基所占百分比,对应红色曲线,右纵坐标为该深度及以下的碱基所占百分比,对应蓝色曲线。4.4 染色体覆盖度统计根据染色体各位点的覆盖深度情况进行作图,若覆盖深度在染色体上的分布比较均匀,则可以认为测序随机性比较好。样品的染色体覆盖深度分布图如下所示: 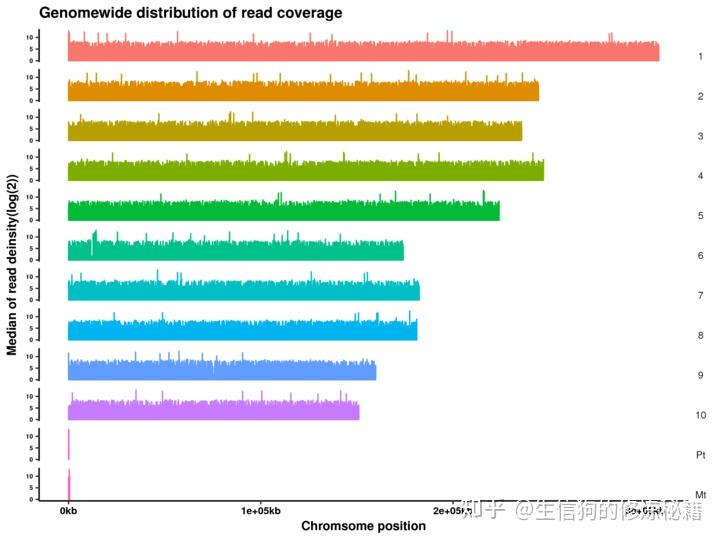 注:横坐标为染色体位置,纵坐标为染色体上对应位置的覆盖深度取对数(log2)得到的值。六、变异检测 注:横坐标为染色体位置,纵坐标为染色体上对应位置的覆盖深度取对数(log2)得到的值。六、变异检测见下一篇文章《WGS全基因组测序数据分析教程(二)》 |
【本文地址】