| 成药性相关小知识(5):血脑屏障通透性 | 您所在的位置:网站首页 › 10π表 › 成药性相关小知识(5):血脑屏障通透性 |
成药性相关小知识(5):血脑屏障通透性
|
概要血脑屏障(BBB)限制一些化合物通透的原因包括∶ P-糖蛋白外排、细胞旁渗透不足以及胞饮作用有限。可以根据化合物的血脑屏障渗透性或脑/血浆分配比来评估其脑暴露。减少氢键数、分子量、P-糖蛋白外排、代谢和血浆蛋白结合,或增加Log P,可以提高化合物的脑暴露。 随着社会老龄化和竞争压力的增大, 中枢神经系统 (central nervous system, CNS) 疾病已成为继心血管疾病之后的第二大疾病[1]。然而, 中枢神经系统 药物研发的成功率却很低, 与心血管疾病药物 20% 的成功率相比, 中枢药物的成功率只有 8%[1]。影响中枢药物研发成功的一个主要因素就是血脑屏障(blood-brain barrier, BBB), 几乎阻挡了 100%的大分 子药物及大于 98%的小分子药物[2]。因此, 除了需要 具有较好的活性和代谢性质及较低的毒性等性质 之外, 中枢神经系统药物还需要克服血脑屏障, 在中枢系统达到足够的暴露量, 这是中枢药物研发成功的关键前提。 1、血脑屏障的基本原理血脑屏障由脑毛细血管内皮及其细胞间的紧密 连接、基底膜、周细胞以及星形胶质细胞等围成的神经胶质膜构成, 其中内皮细胞是血脑屏障的主要结 构, 中枢药物必须透过内皮细胞才能进入脑细胞。除 内皮细胞与星形胶质细胞等形成的物理屏障外, 血 脑屏障还包括各种酶与转运体形成的生化屏障[5]。 血脑屏障通透机制包括被动扩散、主动转运和 外排转运等。被动扩散是小分子药物进入大脑的主 要方式。在正常情况下, 脑毛细血管内皮细胞的有效 孔径为 1.4~1.8 nm, 直径小于 1.8 nm 的小分子可由 被动扩散透过血脑屏障。主动转运是一类需要能量 和载体蛋白参与的逆浓度差、逆电化学梯度的特殊 转运方式。而外排转运系统主要通过 P-糖蛋白 (P-glycoprotein, P-gp) 主动将毒性代谢物和异源性物质 排出, 以维持大脑正常的生理功能[6](图 1)。   图1 脑毛细血管截面示意图及血脑屏障的通透机制[7]2、评价血脑屏障的常用参数2.1 评价血脑屏障的理化参数 图1 脑毛细血管截面示意图及血脑屏障的通透机制[7]2、评价血脑屏障的常用参数2.1 评价血脑屏障的理化参数与非中枢神经系统药物相比, 氢键、脂溶性和分子大小等理化性质较大地影响化合物的血脑屏障通透能力, 如 ① 中枢神经系统药物脂溶性一般较高, 其 LogP 在 2~5 之间[6]; ② 分子质量通常小于 450 Da[8]; ③ 多为中性或弱碱性分子, pKa 在 7.5~10.5 之 间[5]; ④ 较少的氢键供体数目 (HBD), 一般小于 3[8]; ⑤ 较低的氢键结合能力, △LogP 通常小于2[7]; ⑥ 较低的分子极性表面积 (PSA), 一般低于 90 Å2[8]; ⑦ 多为球形分子, 增加支链会降低透过血脑屏障的 能力[8]; ⑧ 分子柔性较低, 可旋转键数目少[8](表 1)。  表 1 中枢药物的常用理化性质参数2.2 评价血脑屏障的实验参数 表 1 中枢药物的常用理化性质参数2.2 评价血脑屏障的实验参数常用评价血脑屏障透过性质的实验参数包括: 脑与血浆中全药浓度的比值 (B/P)、表观渗透系数 (Papp)、外排率 (ER)、游离药物在脑与血浆中比值 (Kp,uu)、游离药物在血浆和脑及脑脊液中的浓度 (Cp,u、Cb,u、CCSF) 等[1](表 2)。  表 2 常用衡量血脑屏障通透性的实验参数. CNS+表示可进 入中枢的化合物, CNS−表示不可进入中枢的化合物3、改善化合物脑通透性的结构修饰策略 表 2 常用衡量血脑屏障通透性的实验参数. CNS+表示可进 入中枢的化合物, CNS−表示不可进入中枢的化合物3、改善化合物脑通透性的结构修饰策略综合考虑中枢药物的特征及药物进出血脑屏障的特点, 通过结构优化可以有效地改善化合物透过 血脑屏障的能力。常用的优化策略包括: 针对被动扩 散的改造——增加脂溶性、减少氢键供体、简化分子、 增加刚性、降低极性表面积、剔除羧基以及前药策略 等; 针对主动运输的改造——将化合物修饰为主动 转运体的底物; 针对外排率较高的化合物——规避 易被外排转运体识别的基团。 3.1 针对被动扩散的改造策略3.1.1 增加脂溶性 研究表明, 脂溶性高的化合物 更易透过血脑屏障, 且能较快地达到分布平衡。因此, 在改善血脑屏障透过性质时, 可以通过引入脂溶性 基团 (如氟、氯)、替换大极性基团等策略增加化合 物的脂溶性。 如化合物 4 (AZD3839) 是 AstraZeneca 公司[12]报道的用于阿尔兹海默症治疗的 BACE1 抑制剂。研 发过程中, 通过引入氟、甲基、二氟甲基等脂溶性基 团增加化合物血脑屏障透过能力。在吲哚环引入氟 后, 嘧啶环 2、6 位分别引入一个甲基得到化合物 2, 其 eLogD 由 0.7 增加到 1.4, 同时, Papp由 3.4×10−6 cm·s−1 提升至 1.9×10−5 cm·s−1 ; 当嘧啶环 2 位引入甲氧基取 代后 (3), 化合物 eLogD 增加到 2.0, 同时 Papp提升至 2.1×10−5 cm·s−1 , 但该化合物甲氧基的存在使得其代 谢稳定性较差; 当嘧啶环 2 位引入二氟甲基后 (4), 其 eLogD 由 0.7 提高至 2.0, 同时 Papp 由 3.4×10−6 cm·s−1 提升至 3.4×10−5 cm·s−1 , 大大增加了化合物的透膜性 质。AstraZeneca 公司通过对化合物活性、Caco-2 细 胞透膜性质、代谢稳定性、hERG 毒性等性质综合衡 量后, 选择了化合物 4 作为临床候选药物, 目前该化合物处于临床 I 期研究中 (图 2)。   图 2 增加脂溶性提高 BACE1 抑制剂的表观渗透系数 图 2 增加脂溶性提高 BACE1 抑制剂的表观渗透系数化合物 (S)-8 (RG1678) 是 Roche 公司[13]报道的第一个选择性 GlyT1抑制剂, 用于精神分裂症的治疗。向化合物5引入氟后, 化合物 6的脂溶性及脑通透性均有一定的提高, 其 cLogP 由 3.92 提高至 4.13, B/P 由 1.10 提升至 1.15。引入极性的吡啶环时, 化合物7的脂溶性下降, cLogP 由 3.92 降为 2.82, B/P 由 1.10 降低至 0.2; 然而, 再次引入脂溶性的氟原子, 获得了活性和脑通透性质均更好的化合物 8。目前, 化合物 (S)-8 已经进入临床 II 期研究, 用于精神分裂症的治疗(表 6)。  表 6 脂溶性对 GlyT1 抑制剂 B/P 的影响 表 6 脂溶性对 GlyT1 抑制剂 B/P 的影响化合物 12 是喹哌嗪类 5-HT3 激动剂, 可调节中 枢的乙酰胆碱释放, 用于神经变性和失调疾病的治 疗[6, 15]。然而大鼠静脉给药后,化合物 12 血脑屏障通透性较差, 其 B/P 值仅为 0.1。通过引入极性较小的 羟甲基后,化合物13的脂溶性大大提高,cLogP 由0.19升高至2.3, B/P 达到20.3。化合物12具有羧基, 在生理条件下, 易在体内以离子状态存在, 难以透过血脑屏障, 通过羟甲基替代羧基, 不仅使化合物的脂溶性有了较大的提高,同时避免了羧酸基团,极大地改善了化合物的血脑屏障通透性 (图 3)。  图 3 增加脂溶性对 5-HT3 激动剂 B/P 的影响 图 3 增加脂溶性对 5-HT3 激动剂 B/P 的影响增加化合物的脂溶性可以有效的改善血脑屏障通透性, 然而也可能对血脑屏障通透性带来负面效应。按照“药动学规则”[7] , 增加化合物的脂溶性往往会增加其在脑中的非特异性结合, 这将会降低脑细胞外液中游离化合物的浓度, 从而降低化合物的活性。因此, 进行脂溶性的结构改造时要注意平衡各项参数, 既要优化化合物的通透性, 又要减少与脑蛋 白的非特异性结合, 提高脑内的药物浓度。 3.1.2 减少氢键供体 中枢药物普遍具有更少的氢 键供体数目, 且许多具有裸露 NH 的化合物具有较为 明显的 P-糖蛋白外排, 故减少化合物氢键供体是中枢药物优化的重要改造策略之一。常用减少氢键供体 的方法包括: 封闭氢键供体、生物电子等排替换氢键 供体及形成分子内氢键等。 化合物 14 是 GlaxoSmithKline 公司[16]报道的 CB2 激动剂先导化合物, 在急性疼痛模型中有较好活 性 (EC50=11 nmol·L−1 ), 但该化合物结构中的吲哚环 含有裸露的 NH, 为 P-糖蛋白底物, 存在较为显著的 外排 (ER = 74), 所以在大鼠脑中的通透性较差, B/P 小于 0.05。直接对吲哚环裸露的 NH 进行 N-甲基化 得到的化合物活性有了较为明显的降低 (EC50 = 654 nmol·L−1)。通过对化合物骨架的修饰, 引入了含有 5-氮杂吲哚环的异构体, 并通过 N-甲基化封闭吲哚 的氢键供体, 得到衍生物 15, 其外排率降低为 2.9, B/P 升高至 1.04, 同时保持了较好的 CB2 激动活性 (EC50=8 nmol·L−1 ) (图 4)  图 4 减少氢键供体对 CB2 激动剂 B/P 的影响 图 4 减少氢键供体对 CB2 激动剂 B/P 的影响化合物 16 是 Lundbeck 公司与中国科学院上海药物研究所[17]通过高通量筛选获得的 GPR139 激动剂, 该化合物 B/P 仅为 0.03。采用生物电子等排策略, 引入亚甲基替换胺基, 减少氢键供体数目, 同时规避了易被P-糖蛋白识别的脲结构,得到衍生物17, 其 B/P 值升高到 2.8,是化合物16的93 倍 (图 5)。  图 5 减少氢键供体对 GPR139 激动剂 B/P 的影响 图 5 减少氢键供体对 GPR139 激动剂 B/P 的影响化合物18是 Valerie 等[18]设计的选择性NK1受体拮抗剂, 可用于镇痛药物的研发。该化合物具有极好的活性和药代动力学性质 (IC50=1.05 nmol·L−1 , F= 50%~60%, t1/2>6 h), 但其血脑屏障通透性较低, B/P 仅为 0.6。化合物19 通过引入N, N-二甲氨基与邻近 的两个酰胺键的 NH 形成了分子内氢键, 提高了血脑 屏障通透性, B/P 达到 6.0, 是 18 的 10 倍, 同时活性 得到保持 (IC50=0.46 nmol·L−1 , 图 6)。 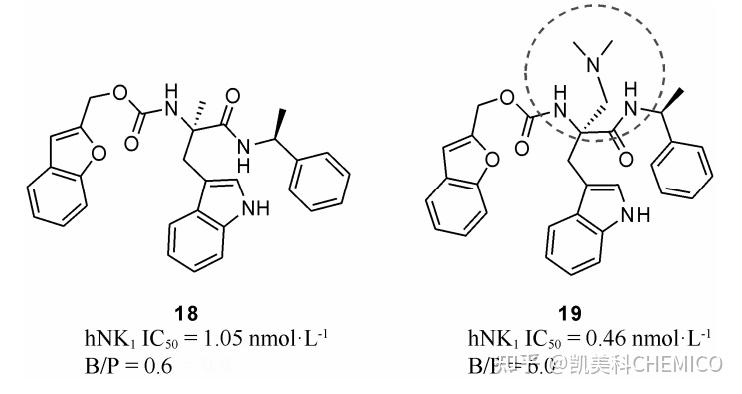 图 6 分子内氢键对 NK1 受体拮抗剂 B/P 的影响 图 6 分子内氢键对 NK1 受体拮抗剂 B/P 的影响3.1.3 简化结构 对化合物结构进行简化, 从而减 小体积和降低相对分子质量, 可以有效改善化合物 的脑通透性, 增加中枢的药物浓度。mGlu4 受体正向 变构调节剂是帕金森症的潜在治疗药物, 但是报道的大部分 mGlu4 受体正向变构调节剂具有类药性、 药代动力学性质或血脑屏障通透性差的不足, 如化 合物 20 具有较好的 mGlu4 受体正向变构调节活性, 但是其脑通透性较差, B/P 不足 0.1; 而结构简化的化 合物 21 和 22 的脑通透性则大大提高, 尤其是化合物 22 的 B/P 值可达到 4.1, 有效地改善了化合物的脑通 透性。Jones 等[19]通过进一步的结构修饰, 最终获得活性和脑通透性均适中的化合物 23 (ML182), 用作 mGlu4 受体正向变构调节剂工具分子, 在氟哌啶醇 介导的肌肉僵直模型中表现出较好的疗效 (图 7)。  图 7 相对分子质量对 mGlu4 正向变构调节剂 B/P 的影响 图 7 相对分子质量对 mGlu4 正向变构调节剂 B/P 的影响化合物 24~27 是 Johnson & Johnson 公司[20]报道 的 γ-分泌酶调控剂。在对其进行 SAR 研究时, 发现 化合物脑通透性呈现较为明显的随分子减小而增大 的趋势。如将 R1 中的三氟甲基苯基简化为苯基时, 化 合物 B/P 值提升至 0.34; 去掉 R 取代基, 化合物 B/P 值进一步提高; 将 R1 位的苯基替换为甲基时, 化合物 的 B/P 值达到 0.88, 获得了更好的脑通透性 (表 8)。  表 8 分子大小对 γ-分泌酶调制剂 B/P 的影响 表 8 分子大小对 γ-分泌酶调制剂 B/P 的影响3.1.4 增加刚性 中枢神经系统药物普遍含有更低 的分子柔性, 因此通过成环等手段增加分子刚性也 是改善化合物血脑屏障通透性的策略之一。化合物 31 (SB-271046) 是第一个进入临床 I 期的 5-HT6 受体 选择性拮抗剂, 但因其血脑屏障通透性较差(B/P = 0.05) 被中止研发。GlaxoSmithKline 公司[21]通过对 其骨架进行修饰形成骈环, 增加了分子刚性, 成环后 化合物 32~34 的 B/P 值均有较为明显的提升, 分别 达到 3.0、2.6 和 0.7, 明显改善了化合物的脑通透性 (图 9)。  图 9 增加分子刚性对 5-HT6 受体选择性拮抗剂 B/P 值的影响 图 9 增加分子刚性对 5-HT6 受体选择性拮抗剂 B/P 值的影响化合物 35 是 Wyeth 公司[22]研发的胍类 BACE1 抑制剂, 其脑通透性较低, B/P 仅为 0.04。将酰胍基替 换为 2-氨基吡啶, 增加了分子刚性, 有效地增加了化 合物的脑通透性, 化合物 36 的 B/P 值升高至 1.7, 是 化合物 35 的 42 倍 (图 10)。  图 10 增加分子刚性对 BACE 抑制剂 B/P 的影响 图 10 增加分子刚性对 BACE 抑制剂 B/P 的影响3.1.5 降低极性表面积 近年来, 分子极性表面积(PSA) 参数引起了人们越来越多的关注[23] , 中枢药 物的分子极性表面积较其他治疗领域药物分子更小。降低化合物的分子极性表面积, 可以有效的增加化合物的血脑屏障通透性。化合物 38 (URMC-099) 是Biofocus公司[24]报道的蛋白激酶3(MLK3) 抑制剂, 可用于帕金森及伴随HIV-1 的认知失调的治疗。在其研发中, 通过降低化合物37的的分子极性表面积获得化合物38 (PSA 从 72 Å2 降低至51Å2 ),有效地增加了化合物的脑通透性, 最大脑浓度 Cmax 由 3736μg·kg−1提高至4685 μg·kg−1 , 同时B/P由0.99提高至1.6 (表 9)。  表 9 分子极性表面积对 MLK3 抑制剂 B/P 的影响 表 9 分子极性表面积对 MLK3 抑制剂 B/P 的影响3.1.6 剔除羧基 含有羧基的药物在体内 pH 条件 下易以离子形式存在,难以透过血脑屏障而发挥药效。因此, 在中枢药物设计时,应尽量规避羧酸基团。 如 Bristol-Myers Squibb 公司[25]开发的第一代 γ-分泌 酶抑制剂 39,由于羧基的存在,使得脑通透性较差, 其 B/P 值仅为 0.2,在临床 I 期试验中被终止。他们在其基础上剔除了羧酸基团,研发出第二代 γ-分泌酶抑制剂 40 (BMS-708163), 其 B/P 值获得了较大的突破, 由 0.2 提高至 2.4,且活性也获得一定的提升。目前,该化合物已经进行临床 II 期试验,用于阿尔兹海默症的治疗 (图 11)。  图 11 剔除羧酸基因对 γ-分泌酶 B/P 的影响 图 11 剔除羧酸基因对 γ-分泌酶 B/P 的影响3.1.7 前药策略 通过对化合物进行前药修饰是中 枢药物研发的常用策略之一。前药修饰策略通常包括酯化、酰化、酰胺化和拼合等[26]。经典的前药包括 镇痛药吗啡的乙酰化药物海洛因、神经递质左旋多巴 的酯化药物左旋多巴甲酯和左旋多巴乙酯、赛奥芬的 乙酰化前药乙酰化赛奥芬及酯化前药醋托酚等[27]。 3.2 修饰为主动转运体底物(略) 3.3 减少外排外排使得中枢药物研发变得更加复杂。中枢的外排转运体包括 P-糖蛋白 (P-gp) 转运体、多药耐药蛋白 (MRP) 转运体、乳腺癌耐药蛋白 (BCRP)转运体、有机阴离子转运体 (OAT) 和谷氨酸转运体 (EAAT) 等, 其中P-糖蛋白是最主要的外排转运体。为减少化合物的P-糖蛋白外排, 化合物不仅要符合非P-糖蛋白底物的一般理化特征 (如氮氧数目之和 不大于 4, 相对分子质量小于 400, pKa 小于 8 等[10]), 还应尽量避开易被 P-糖蛋白识别的基团, 如含有NH 的杂环、磺胺基、脲等[30]。 3.3.1 含有NH 杂环的结构优化 化合物 52 是 Merck 公司[31]发现的OX1/OX2 双重抑制剂的先导化合物, 用于睡眠失调治疗药物的研发。尽管化合物52具有较好的生物活性 (hOX1 Ki=28nmol·L−1, hOX2 Ki=1nmol·L−1 ), 但是对其 SAR 研究发现化合物52和53都是P-糖蛋白的底物, 其外排率高达 6.8 和 13。他们通过 N-甲基化,封闭了P-糖蛋白的识别的杂环NH,使化合物54和55外排明显下降(ER=2),提高了脑内化合物含量 (表10)。  表 10 N-甲基化对 OX1/OX2 双重抑制剂外排率 (ER) 的影响 表 10 N-甲基化对 OX1/OX2 双重抑制剂外排率 (ER) 的影响3.3.2 磺胺基的结构改造 化合物 56 是 Glaxo Smith Kline 公司[32]报道的AMPA受体正向变构调节剂,用于阿尔兹海默症、帕金森症、抑郁症和注意力缺陷多 动症等的治疗。口服后在体内的 B/P 值仅为 0.1。研 究发现该化合物是 P-糖蛋白的底物 (ER = 5.8),通 过对磺胺进行 N-甲基化,化合物 57 的外排率降低至 3.2,其 B/P 由 0.1 提高到 0.4; 引入 2-氟吡啶环后,化 合物不再是 P-糖蛋白底物,外排率降低为 1.1, B/P 升 高至 2.1,有效地提高了脑内药物浓度 (图 16)。  图 16 规避磺胺基对 AMPA 受体正向变构调节剂 B/P 和 ER 的影响4、ACD/Percepta软件的血脑屏障通透性预测 图 16 规避磺胺基对 AMPA 受体正向变构调节剂 B/P 和 ER 的影响4、ACD/Percepta软件的血脑屏障通透性预测在药物发现早期,通过软件对化合物的脑通透性进行预测是比较有指导作用的。影响化合物脑通透性的因素很多(如 P-gp转运外排、被动扩散、摄取、血浆蛋白结合、脑组织结合、代谢清除),因此这对计算机预测模型是一个复杂的挑战。 ACD/Percepta软件中内置了专属的BBB预测模型,不仅可预测化合物的理化性质(如上文2.1所述,logP、pKa、PSA、HBD等能帮助判断化合物的血脑屏障通透性),还可以直接预测以下相关参数: LogPS——Rate of brain penetration LogBB——Extent of brain penetration Log (PS * fu, brain)—Brain/plasma equilibration rate软件还会把测试化合物与一组中枢神经药物、一组外周药物进行比较,通过散点图的形式显示测试化合物所处的位置。如下图所示:  如上图所示,ACD/Percepta软件中的BBB预测界面显示了化合物的理化性质,如LogP、pKa、血浆游离分数(Fraction unbound in plasma),基于此主要的理化性质结果再模拟其血脑通透参数(logPS、logBB、log(PS*fu,brain))。用户可对这些理化参数进行修改,比如输入实验值,再进行模拟,以获得更准确的结果。 在另外,软件还会预测化合物是否会被某些(吸收/外排)转运体介导发生转运,从而影响化合物血脑屏障穿透性。除了可预测化合物是否为P-gp外排转运体底物外,还包括以下主动吸收转运体: Amino-acid transport systems (System A, L, y+, x-)Glucose carrier GLUT1Organic cation/carnitine transporters (OCTN)Organic anion transporting polypeptide (OATP)Various other carriers specific for nucleotides and other endogenous substances.如果预测结果提示化合物是某个转运体的底物,用户得重新考虑以上的BBB预测结果。 如需了解ACD/Percepta软件更多信息,请联系:广州凯美科信息技术有限公司 http://www.chemico.com.cn 产品经理:胡经理 联系电话:18680221948 联系邮箱:[email protected] 本文章内容主要摘自以下文献: 药学学报Acta Pharmaceutica Sinica 2014, 49 (6): 789−799 先导化合物结构优化策略 (四)—— 改善化合物的血脑屏障通透性 参考文献 [1] Di L, Rong H, Feng B. Demystifying brain penetration in central nervous system drug discovery [J]. J Med Chem, 2013, 56: 2−12. [2] Gabathuler R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases [J]. Neurobiol Dis, 2010, 37: 48−57. [3] Palmer AM, Alavijeh MS. Translational CNS medicines research [J]. Drug Discov Today, 2012, 17: 1068 −1078. [4] Hitchcock SA, Pennington LD. Structure-brain exposure relationships [J]. J Med Chem, 2006, 49: 7559 −7583. [5] Kerns E, Di L. Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization [M]. Beijing: Academic Press, 2008: 122 −135, 311−328. [6] Ghose AK, Herbertz T, Hudkins RL, et al. Knowledgebased, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery [J]. ACS Chem Neurosci, 2012, 3: 50−68. [7] Mensch J, Oyarzabal J, Mackie C, et al. In vivo, in vitro and in silico methods for small molecule transfer across the BBB [J]. J Pharm Sci, 2009, 98: 4429−4468. [8] Swahn BM, Kolmodin K, Karlstrom S, et al. Design and synthesis of β-site amyloid precursor protein cleaving enzyme (BACE1) inhibitors with in vivo brain reduction of β-amyloid peptides [J]. J Med Chem, 2012, 55: 9346 −9361. [9] Pinard E, Alanine A, Alberati D, et al. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin- 2-yl) piperazin-1-yl][5-methanesulfonyl-2-((S)-2, 2, 2-trifluoro- 1-methylethoxy) phenyl] methanone (RG1678), a promising novel medicine to treat schizophrenia [J]. J Med Chem, 2010, 53: 4603−4614. [10] Campiani G, Morelli E, Gemma S, et al. Pyrroloquinoxaline derivatives as high-affinity and selective 5-HT 3 receptor agonists: synthesis, further structure-activity relationships, and biological studies [J]. J Med Chem, 1999, 42: 4362 −4379. [11] Giblin GM, Billinton A, Briggs M, et al. Discovery of 1-[4- (3-chlorophenylamino)-1-methyl-1H-pyrrolo [3, 2-c] pyridin- 7-yl]-1-morpholin-4-ylmethanone (GSK554418A), a brain penetrant 5-azaindole CB2 agonist for the treatment of chronic pain [J]. J Med Chem, 2009, 52: 5785 −5788. [12] Shi F, Shen JK, Chen D, et al. Discovery and SAR of a series of agonists at orphan G protein-coupled receptor 139 [J]. ACS Med Chem Lett, 2011, 2: 303−306. [13] Ashwood VA, Field MJ, Horwell DC, et al. Utilization of an intramolecular hydrogen bond to increase the CNS penetration of an NK1 receptor antagonist [J]. J Med Chem, 2001, 44: 2276−2285. [14] Jones CK, Engers DW, Thompson AD, et al. Discovery,synthesis, and structure-activity relationship development of a series of N-4-(2, 5-dioxopyrrolidin-1-yl) phenylpicolinamides (VU0400195, ML182): characterization of a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4) with oral efficacy in an antiparkinsonian animal model [J]. J Med Chem, 2011, 54: 7639 −7647. [15] Bischoff F, Berthelot D, De Cleyn M, et al. Design and synthesis of a novel series of bicyclic heterocycles as potent γ-secretase modulators [J]. J Med Chem, 2012, 55: 9089 − 9106. [16] Ahmed M, Briggs MA, Bromidge SM, et al. Bicyclic heteroarylpiperazines as selective brain penetrant 5-HT 6 receptor antagonists [J]. Bioorg Med Chem Lett, 2005, 15: 4867−4871. [17] Malamas MS, Barnes K, Hui Y, et al. Novel pyrrolyl 2- aminopyridines as potent and selective human β-secretase (BACE1) inhibitors [J]. Bioorg Med Chem Lett, 2010, 20: 2068−2073. [18] Clark DE. What has polar surface area ever done for drug discovery? [J]. Fut Med Chem, 2011, 3: 469 −484. [19] Goodfellow VS, Loweth CJ, Ravula SB, et al. Discovery, synthesis, and characterization of an orally bioavailable, brain penetrant inhibitor of mixed lineage kinase 3 [J]. J Med Chem, 2013, 56: 8032−8048. [20] Gillman KW, Starrett JE Jr, Parker MF, et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable γ-secretase inhibitor [J]. ACS Med Chem Lett, 2010, 1: 120−124 [21] Sozio P, Cerasa LS, Abbadessa A, et al. Designing prodrugs for the treatment of Parkinson’s disease [J]. Expert Opin Drug Discov, 2012, 7: 385−406. [22] Hitchcock SA. Structural modifications that alter the Pglycoprotein efflux properties of compounds [J]. J Med Chem, 2012, 55: 4877−4895. [23] Bergman JM, Roecker AJ, Mercer SP, et al. Proline bisamides as potent dual orexin receptor antagonists [J]. Bioorg Med Chem Lett, 2008, 18: 1425 −1430. [24] Ward SE, Harries M, Aldegheri L, et al. Discovery of N- [(2S)-5-(6-fluoro-3-pyridinyl)-2, 3-dihydro-1H-inden-2-yl]-2- propanesulfonamide, a novel clinical AMPA receptor positive modulator [J]. J Med Chem, 2010, 53: 5801 −5812. |
【本文地址】